AMD Mimickers: When to Suspect Macular Dystrophy
Some presentations are different than they appeared at first glance. Here's how to spot the differences.
By Sara Weidmayer, OD
Release Date: June 2017
Expiration Date: June 15, 2020
Goal Statement: Since many maculopathies appear quite similar clinically, this article is designed to educate practitioners about similarities and differences between several types of maculopathies, as well as explain how imaging devices and other tools can be used to differentiate these entities.
Faculty/Editorial Board: Sara Weidmayer, OD
Credit Statement: This course is COPE approved for 2 hours of CE credit. Course ID is 53929-PS. Check with your local state licensing board to see if this counts toward your CE requirement for relicensure.
Disclosure Statements:
Authors: The author has no relationships to disclose.
Editorial staff: Jack Persico, Rebecca Hepp, William Kekevian, Michael Riviello and Michael Iannucci all have no relationships to disclose.
Age-related macular degeneration (AMD) is a progressive disease of the central retina and the leading cause of blindness in the developed world.1 While drusen are the trademark finding, AMD may present with any combination of drusen, retinal pigment epithelial (RPE) changes, geographic atrophy (GA) and choroidal neovascularization (CNV).1 Given this variety of clinical presentations, several other diseases look similar to AMD, such as macular dystrophies. Here, we will explore how to differentiate AMD from some of the more commonly seen macular dystrophies so we can appropriately diagnose and manage the right disease and provide accurate prognostic information to our patients.
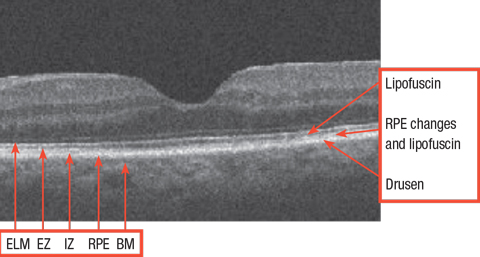 |
| Normal retina with relevant structures noted. ELM: external limiting membrane. EZ: ellipsoid zone. IZ: interdigitation zone. RPE: retinal pigment epithelium. BM: Bruch’s membrane. Click image to enlarge. |
Clinical Features of AMD
The best way to detect a counterfeit dollar bill is not to know all of the possible fraudulent features of a counterfeit, but rather, to know the details of a true dollar bill. Similarly, the best way to detect AMD mimickers is to understand the features of AMD itself.
Drusen, the hallmark of AMD, are small extracellular byproduct deposits that accumulate posterior to the RPE, between the RPE and Bruch’s membrane.1,2 They appear yellowish and vary from small (<63µm) to large (>125µm).1,3 While small drusen may appear simply due to aging and do not necessarily indicate early AMD, medium drusen without pigmentary changes fits the criteria for early AMD. Intermediate stage AMD includes any large drusen, or any macular pigmentary changes associated with medium or large drusen. Late AMD shows GA, CNV or disciform scarring (Case 1).1,4
The pigmentary changes seen in AMD occur as a result of RPE degeneration or migration.1 Several factors contribute to the degeneration of RPE cells, including oxidative stress and inflammation.1 Oxidative stress may occur due to the RPE’s physiologic role in outer segment phagocytosis and environmental factors including chemical oxidative stress related to smoking, high fat diets and photo-oxidative stress from sunlight exposure. These oxidative stressors trigger an immune response, resulting in both acute and chronic regional inflammation, ultimately injuring the tissue.5 RPE degeneration clinically appears as areas of RPE mottling or atrophy; migratory changes may look like pigment clumps, typically at the level of the RPE, but it is not uncommon to see pigment migration into the outer, or even more anterior, layers of the retina; this is particularly evident on optical coherence tomography (OCT). GA shows larger, defined patches of choriocapillaris, RPE and subsequently outer retinal atrophy (Case 2).1
Any area of disruption in the Bruch’s membrane-RPE complex, including drusen, RPE mottling or atrophy, can compromise the blood-retina barrier and predispose it to the development of CNV. This condition is caused by new blood vessels that form from the choriocapillaris, through Bruch’s membrane and into the sub-RPE or subretinal space; exudation of blood and plasma from CNV causes tissue disruption and later leads to the fibrovascular scar formation that is typical in exudative AMD. CNV may develop in patients with macular dystrophies, but this is infrequent. Fortunately, CNV associated with macular dystrophies generally has a better prognosis than CNV associated with AMD.1
Symptoms typical of AMD are progressive central visual acuity loss, central scotomas and metamorphopsia. Slowed dark adaptation is common in patients with AMD, and patients may be symptomatic for this before there is any loss of central visual acuity.1
Though AMD diagnosis seems straightforward enough, mimickers can trip ODs up. Macular dystrophies in particular have considerable overlap with AMD in the clinical features and symptoms. Luckily, today’s diagnostic tools provide the enhanced ability to detect and differentiate imposters.
| Table 1. How Dystrophies Compare with AMD | |||
| Dystrophy | Similarities with AMD | Differences from AMD | Helpful Ancillary Studies |
| ML | • Drusen • Possible GA or CNV | • Earlier onset (3rd-4th decade) • Likely (+)family history (autosomal dominant) • Smaller radial macular drusen, larger drusen around disc; confluent drusen later on | • Large drusen may hyper-AF (unlike drusen in AMD) |
| Stargardt’s, fundus flavimaculatus | • Yellowish flecks • Possible GA | • Typically early onset (<3rd decade), but rarely can have late onset (>50 years) • Flecks are irregularly shaped and typically extend beyond macula • Flecks may spare the macula • Rarely develops CNV • Likely (+) family history (autosomal recessive) | • Yellowish flecks intensely hyper-AF on FAF and may have surrounding hypo-AF halo • OCT shows flecks as RPE thickening (not sub-RPE as with drusen) |
| Butterfly pattern dystrophy | • Pigmentary changes • Possible GA or CNV | • Mid-life onset (20-50 yrs) • Likely (+) family history • Butterfly pattern of pigmentary changes including lipofuscin | • Butterfly pattern of pigmentary changes on FAF, hyper-AF if lipofuscin present |
| AFVM | • Yellowish macular lesion • Atrophic changes in later stages • Possible CNV | • Mid-life onset (20-50 yrs) • No drusen • Vitelliform lesion is usually solitary | • Vitelliform lesion is anterior to RPE (between RPE and retina) with intense hyper-AF on FAF |
| BVMD | • Yellowish macular lesion • Atrophic changes in later stages | • Earlier onset (<40 yrs) • No drusen • Likely (+) family history (autosomal dominant) • Associated with hyperopia • Rarely develops CNV | • Vitelliform lesion is anterior to RPE (between RPE and retina) with intense hyper-AF on FAF |
| CACD | • Looks like GA | • Typically earlier onset (5th-6th decade), can be later (>55 years) • Usually no drusen • Likely (+) family history (autosomal dominant) • Multiple oval atrophic patches | • Very sharply demarcated hypo-AF atrophic patches on FAF |
| Cone dystrophy | • Pigmentary changes • Possible GA | • Most often earlier onset (30-60 years) • Possible (+) family history • No drusen • Possible peripheral bone-spicule pigmentation and/or temporal optic disc pallor | • Atrophic changes on FAF • Outer retinal thinning on OCT with loss of hyperreflective outer bands (EZ, IZ) • Significant decrease in photopic response on ERG • Color testing abnormal |
Ancillary Tools
Color testing. This is a simple way to get a glimpse at the patient’s central cone function. Color discrimination can be reduced in AMD, but usually not by any substantial amount. Color vision is generally more notably reduced in macular dystrophies, particularly those specifically affecting cone function, such as cone dystrophy.1 Serial color vision testing may assist in monitoring functional disease progression in any maculopathy.
| Case 1. Drusen in Dry AMD Patient |
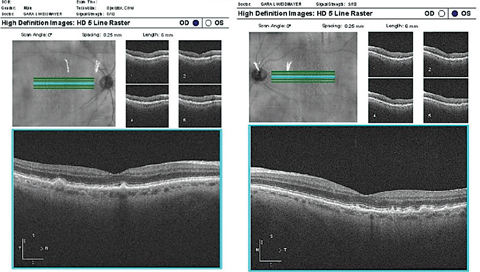 |
| These OCT images show typical drusen in dry AMD. Notice that the drusen are under the RPE. Click image to enlarge. |
Visual fields. While AMD certainly will show variable degrees of central visual field (VF) loss, and VFs aren’t typically routinely repeated in AMD patients, some macular dystrophies in their early stages may cause patient symptoms before clinical signs. Especially in patients with complaints of qualitative vision loss but an unremarkable fundus, a central VF (such as 10-2) or microperimetry may aid in the detection and monitoring of functional progression of early macular dysfunction more precisely than standard high-contrast acuity measurements. Both mesoptic and scotopic microperimetry can be performed, and the functional sensitivity results may be correlated to—and tracked along with—the structural findings seen simultaneously on OCT.6
Fundus autofluorescence (FAF). This can serve as one of the clinician’s best tools to help differentiate AMD from macular dystrophies. On FAF, there is normally a low level of autofluorescence, which is derived from the autofluorescence of lipofuscin that is normally found at the level of the RPE.1 These cells phagocytose the outer segment of photoreceptors as they are shed during the normal visual cycle; the phagocytosed material is stored in liposomes, which subsequently form lipofuscin that remains stored in the RPE.7 Lipofuscin precursors can also accumulate between the RPE and retina, and these precursors also autofluoresce.1 Relative to the normal background autofluorescence, any areas of excess lipofuscin accumulation will appear as hyperautofluorescent (hyper-AF) on FAF, and funduscopically appears as a yellowish deposit. Conversely, areas of photoreceptor or RPE atrophy, thus with less lipofuscin, will appear hypo-autofluorescent (hypo-AF) or dark. There may be a junctional ring of hyper-AF around areas of RPE atrophy; this indicates the RPE is likely metabolically stressed in that zone, burdened with an abundance of shed outer segments and is generally indicative of impending atrophy in that area.7
Except for large or confluent drusen, which may be somewhat hyper-AF, drusen associated with AMD tend to be relatively isoautofluorescent (iso-AF), uniform with the low level of background autofluorescence.1 While AMD may have some associated lipofuscin accumulation and may show variable autofluorescence patterns based on the clinical phenotype, specific macular dystrophies often show fairly characteristic distribution patterns of FAF findings. Also, many macular dystrophies begin earlier in life, so end up with higher amounts of lipofuscin accumulation over time, which ultimately intensifies the hyper-AF signal.1
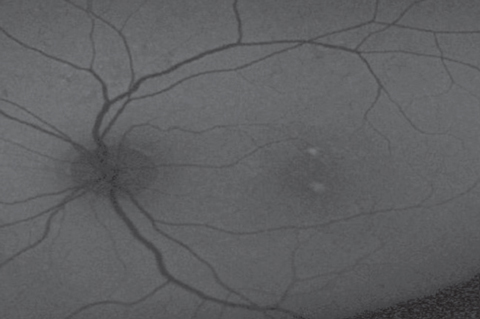
| Case 2. Geographic Atrophy |
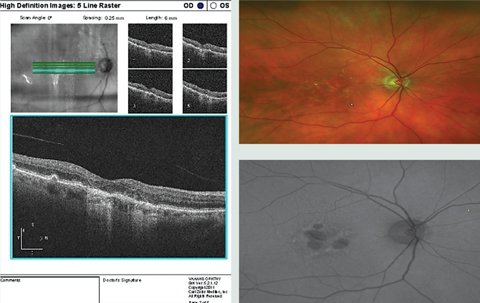 |
| Notice the hypo-AF on FAF associated with the GA, and some patchy surrounding hyperf-AF surrounding the GA in the OCT, color photo and FAF image of this patient with AMD with GA and drusen. On OCT, the obvious drusen is posterior to the RPE, as expected, and the atrophy involves the RPE and outer retina. Click image to enlarge. |
OCT. The anatomic structure of the retina-to-choroid complex can be readily visualized using OCT (Figure 1). In differentiating AMD from mimickers, this offers extremely helpful additional information. Key points to note:
1. Drusen form posterior to the RPE, between the RPE and Bruch’s membrane.
2. RPE changes are just that: RPE changes. RPE clumping or thickening can be visualized with OCT. RPE migration, often into the outer retina, can occur with AMD.
3. Lipofuscin or lipofuscin precursors are either within the RPE, or accumulate anterior to the RPE, between the RPE and retina.
Electroretinogram (ERG) can also help differentiate AMD from its mimickers. It may not be readily available to the primary eye care provider, but ERG may prove helpful in a number of these cases (particularly for deciphering cone dystrophy where there will be an obviously reduced photopic response).1 As in any situation, when in doubt, investigate with further ancillary studies or refer the patient, as appropriate.
Family History
Clearly known environmental risk factors exist for AMD, such as ultraviolet exposure and smoking, and though there is some genetic contribution and AMD may run in families, the bulk of cases are sporadic.1 Macular dystrophies, on the other hand, are largely genetic.1 There can be variable expression and penetrance of macular dystrophies, but remember, relatively symmetric retinal findings suggest heredity, and by-and-large a family history will likely be able to be elicited in macular dystrophies. Referral for genetic testing may be quite helpful for patients with macular dystrophy, certainly for diagnosis, but likely also for the purposes of disease prognosis, and genetic counseling.
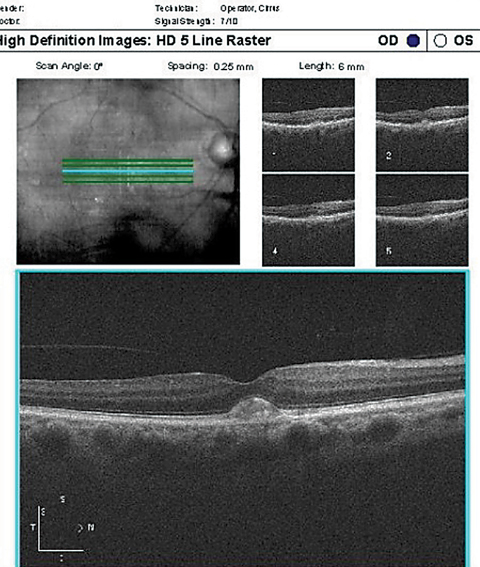
| Case 3. Adult-onset Vitelliform Dystrophy |
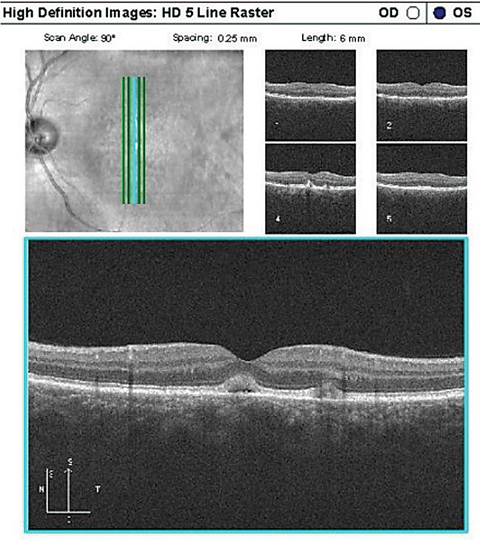 |
 |
| On top, OCT in an AFVM patient showing two lesions with vitelliform material anterior to the RPE, the larger (more central) of the two with an underlying lucent space. On bottom, FAF of the same patient demonstrating the two hyper-AF vitelliform lesions. Click top image to enlarge. |
Types of Dystrophies
Malattia leventinese (ML) (also known as Doyne honeycomb dystrophy or sometimes, non-specifically, as dominant drusen or familial drusen), is characterized by radial drusenoid deposits of varying sizes around the macula and optic nerve that over time can coalesce into large drusenoid plaques.1,2 These
deposits are sub-RPE, as with drusen in AMD, but unlike AMD, the smaller drusen tend to be more elongated and orient radially temporal to the macula, and the larger round drusen are in the perimacula and around the disc. Also unlike drusen in AMD, in some cases the larger drusen in ML tend to hyper-AF (the smaller drusen are iso-AF as generally expected for drusen).2 ML is an autosomal-dominant disease that starts typically in the third to fourth decade, with more rapid progression in the mid-40s; however, there is wide variability in both onset and severity of disease.1,2 Symptoms may include loss of central acuity, central metamorphopsia or scotoma, photosensitivity or color vision decline. These changes occur due to pigmentary and atrophic changes associated with confluent drusen, which can then lead to the development of CNV.1,2
Fundus flavimaculatus (a variation of Stargardt’s disease), an autosomal-recessive dystrophy, usually develops before the third decade of life (Stargardt’s), but rarely can manifest in later years after age 50, fundus flavimaculatus, somewhat mimicking AMD.1,8-10 It shows yellowish flecks of lipofuscin accumulation on clinical exam, similar to drusen but more irregularly shaped and are at the level of the RPE.8,10 The flecks may vary in size and shape and often are fish-shaped, and generally extend beyond and even may spare the central macula.1,8,11 These flecks intensely hyper-AF on FAF, and may have a surrounding hypo-AF ring.1 GA may develop, but CNV is rare; however, CNV in fundus flavimaculatus carries a poor prognosis.11 The juvenile form may begin with symptoms of acuity decline or a reduced sense of color discrimination that seem out of proportion to the clinical exam, where the patient may initially only have a blunted foveal light reflex.8 The juvenile form becomes more visually devastating, while the adult-onset form usually has more widespread flecks but more slowly progressive disease.8,9
Pattern dystrophies include butterfly-shaped pattern dystrophy, adult-onset foveomacular vitelliform dystrophy (AFVM) and others.12 The pattern dystrophies are generally symmetric and include lipofuscin deposits or various patterns of pigmentary changes or atrophy. However, pattern dystrophies can vary substantially within families and even within one patient from eye to eye, and can even shift from one pattern to another over time.1,12 These generally develop mid-life with mild symptoms early on.1,12
Butterfly-shaped pattern dystrophy is a progressive disorder that appears in earlier mid-life with central RPE pigmentary changes and lipofuscin accumulation, often surrounded by adjacent atrophic changes, usually in a patchy radial or butterfly-wing pattern; these RPE changes and atrophy lead to photoreceptor loss with time.1,12,13 Patients are usually asymptomatic at diagnosis and usually progress slowly to mild or moderate acuity loss (unless GA or CNV ensue), while maintaining normal color vision and dark adaptation.12,13
AFVM is classified as a pattern dystrophy, but predominantly manifests early in the disease with vitelliform (yellow egg yolk-like) rather than pigmentary changes. The foveal vitelliform lesion is made of hyperreflective lipofuscin material and other debris between the retina and RPE.12,14 These vitelliform lesions are generally solitary but may be multifocal, are usually symmetric bilaterally and intensely hyper-AF on FAF.1,3,11 Symptoms with AFVM generally develop mid-life and are often reported as only mild visual acuity changes (Cases 3 and 4).1
Similarly, Best vitelliform macular dystrophy (BVMD) begins with only mild foveal pigmentary changes, then evolves to also show a foveal vitelliform lesion that becomes quite large (at least one disc diameter).9 With time, the vitelliform material begins to clump, settle inferiorly and reabsorb, and later leads to chorioretinal atrophy; this evolution in BVMD is not as typical or as dramatic as is seen in AFVM.9,11 However, the vitelliform material can still reabsorb as RPE atrophy develops in AFVM, as well, which can lead to a lucid or optically clear space on OCT within or under the vitelliform lesion.12 In both BVMD and AFVM, the vitelliform lesion develops between the RPE and retina, which is a helpful differentiator from drusen on OCT and shows intense hyper-AF on FAF, whereas associated atrophy would show hypo-AF.15 CNV is possible with AFVM and BVMD, particularly during the reabsorption phase.14 BVMD is autosomal-dominant, but with variable penetrance and expression; central vision loss ensues typically before age 40, but ranges widely.1,9 Interestingly, BVMD is often associated with hyperopia, which might be a helpful clinical consideration.1
| Case 4. Vitelliform Lesion |
 |
| A typical, solitary vitelliform lesion in a patient with AFVM. Notice the vitelliform material is anterior to the RPE. Click image to enlarge. |
Central areolar choroidal dystrophy (CACD) is another autosomal dominant retinal dystrophy.1,9 Visual symptoms—due to central pigmentary changes then eventual atrophy—usually start in the fourth to fifth decades, but up to one-third can begin later in life (>55 years), mimicking AMD.1 CACD may start with subtle pigmentary changes, but the patches of RPE, choriocapillaris and outer retinal atrophy that develop are very sharply demarcated ovals, which expand to coalesce with one another; unlike AMD, CACD rarely has associated drusen.1,12 Patients may experience photophobia along with visual decline.1
Cone dystrophy is a disease of progressive cone dysfunction, which may be inheritable or sporatic.11,16 Signs and symptoms associated with cone dystrophy can develop at any age, and though it is less common to first manifest later in life (>60 years), late-onset cone dystrophy does occur and is often initially associated with central or paracentral vision loss, color vision dysfunction or photophobia without any clear funduscopic findings to substantiate the patient’s symptoms; this can make the initial clinical diagnosis challenging.16 Later in the disease, cone dystrophy may present with a subtle foveal granularity, pigmentary changes, a bull’s-eye appearance, or frank atrophy.1,11,16 Unlike AMD, patients with late-onset cone dystrophy may show peripheral bone-spicule pigmentary changes, particularly with cone-rod dystrophy, and rarely, temporal optic disc pallor.1,11 When retinal findings appear normal or nonspecific, ancillary testing can assist: early atrophic changes may be detectable on FAF, and OCT will show outer retinal thinning with loss of the ellipsoid line due to photoreceptor atrophy, and loss of the interdigitation zone between the photoreceptors and RPE complex.1,17 ERG shows a significant reduction or even absence in photopic response and likely a reduced scotopic response, as well, as many with cone dystrophy also develop rod dysfunction with time, referred to as cone-rod dystrophy.11 This is associated with nyctalopia due to rod dysfunction. ERG changes are evident before symptoms and signs develop, so can be tremendously helpful in these patients.9,16
Many macular dystrophies share features with AMD, such as drusen or drusenoid deposits, pigmentary changes or atrophy.
When patients present with decline in acuity or color vision, or if what looks like AMD doesn’t fit the bill, be sure to further investigate. Color vision testing is simple, yet quite helpful, particularly with cone dystrophy. If a patient reports qualitatively reduced visual function, even if not measurable in office or if the fundus looks normal, get photos with FAF. Central visual field testing may also help lead to early detection of macular disease. OCT can offer substantial additional structural information to differentiate AMD from mimickers. And, as always: when it doubt, refer the patient to a medical retina or retinal dystrophy specialist; use their expertise to the benefit of your patients.
Dr. Weidmayer practices at the VA Ann Arbor Healthcare System in Ann Arbor, MI. She is also a clinical instructor for the University of Michigan Department of Ophthalmology and Visual Sciences.
1. Saksens NT, Fleckenstein M, Schmitz-Valckenberg S, et al. Macular dystrophies mimicking age-related macular degeneration. Progress in Retinal and Eye Research. 2014;39:23-57. |